clavibacter
Searching for Clavibacter michiganensis michiganensis (Cmm) specific sequences


After extracting the specific Clavibacter michiganensis michiganensis (Cmm) reads
from all the libraries, I need to evaluate if these sequences are truly from
Cmm. The reads separation was made by the kraken2 algorithm using its database.
The main issue is if this sequences are really from Cmm and not from one of the
other subspecies fo Clavibacter michiganensis.
The mock microbiome database
I downloaded a set of genomes of the michiganensis subspecies from the NCBI:
| Cm subspecies | NCBI-link | Number of genomes |
|---|---|---|
| capsici | link | 4 |
| tessellarius | link | 3 |
| insidiosus | link | 7 |
| nebraskensis | link | 12 |
| sepedonicus | link | 3 |
The reference genomes are located inside the data/reference/clavi-genomes/
folder of the repository.
I also used a reference genome of Cmm
Correction of the genomes files
With this amassed database, I faced a problem. Each one of them has its own header/label nomenclature for each read/contig/scaffold. For example, the reference Cmm genome has this headers:
$ grep '>' cmm-contigs.fa
>NC_009480.1
>NC_009478.1
>NC_009479.1
To change this, I created a little program called changing-headers.sh which takes the headers of a fasta file and change them for a desired one. It is important to highlight that all the headers are going to be the same.
$ cat changing-headers.sh
#!/bin/sh
# This program is to change the headers of a fasta file in order for all the
#headers to be the same for the entire file
# This is useful when the user want to concatenate all the genomes from a
#database and want to know from which of the initial genomes is the
#best match
#This program ask from the user to specify 2 things: 1) the location
#of the fasta file and 2) the suffix that all the identifiers are going
#to be changed for.
gem=$1 #Location to the genome in fasta format
sufx=$2 #The new header without the '>' symbol at the begginng
#CREATE A .TXT FILE TO STORE THE ORIGINAL HEADERS
grep '>' $gem > heads.txt
cp $gem $sufx.fna
#REPLACE ALL THE HEADERS FOR THE SUFFIX NAME
cat heads.txt | while read line; do sed "s/$line/>$sufx/" $sufx.fna > temp.txt; mv temp.txt $sufx.fna ;done
#REMOVING TEMP FILES
rm heads.txt
I will run it in a new directory called trim-c-genomes:
$ mkdir trim-c-genomes
$ sh changing-headers.sh ../cmm/cmm-contigs.fa Cmm
$ grep '>' Cmm.fna
>Cmm
>Cmm
>Cmm
With this result, I will apply this to all the Clavibacter genomes that I downloaded previously:
$ ls clavi-genomes/
Clavibacter_michiganensis_subsp_capsici_1101.fna
Clavibacter_michiganensis_subsp_capsici_1106.fna
Clavibacter_michiganensis_subsp_capsici_1207.fna
Clavibacter_michiganensis_subsp_capsici_PF008.fna
Clavibacter_michiganensis_subsp_insidiosus_ATCC_10253.fna
Clavibacter_michiganensis_subsp_insidiosus_CFBP_1195.fna
Clavibacter_michiganensis_subsp_insidiosus_CFBP_2404.fna
Clavibacter_michiganensis_subsp_insidiosus_CFBP_6488.fna
Clavibacter_michiganensis_subsp_insidiosus_LMG_3663.fna
Clavibacter_michiganensis_subsp_insidiosus_R1-1.fna
Clavibacter_michiganensis_subsp_insidiosus_R1-3.fna
Clavibacter_michiganensis_subsp_nebraskensis_419B.fna
Clavibacter_michiganensis_subsp_nebraskensis_44.fna
Clavibacter_michiganensis_subsp_nebraskensis_61-1.fna
Clavibacter_michiganensis_subsp_nebraskensis_7580.fna
Clavibacter_michiganensis_subsp_nebraskensis_A6096.fna
Clavibacter_michiganensis_subsp_nebraskensis_CFBP_7577.fna
Clavibacter_michiganensis_subsp_nebraskensis_CIBA.fna
Clavibacter_michiganensis_subsp_nebraskensis_DOAB_395.fna
Clavibacter_michiganensis_subsp_nebraskensis_DOAB_397.fna
Clavibacter_michiganensis_subsp_nebraskensis_HF4.fna
Clavibacter_michiganensis_subsp_nebraskensis_NCPPB_2581.fna
Clavibacter_michiganensis_subsp_nebraskensis_SL1.fna
Clavibacter_michiganensis_subsp_sepedonicus_ATCC33113.fna
Clavibacter_michiganensis_subsp_sepedonicus_CFIA-Cs3N.fna
Clavibacter_michiganensis_subsp_sepedonicus_CFIA-CsR14.fna
Clavibacter_michiganensis_subsp_tessellarius_ATCC_33566.fna
Clavibacter_michiganensis_subsp_tessellarius_DOAB_609.fna
The line of code that I use to change all the headers from all the files, is the next one:
$ cd trim-c-genomes
$ ls clavi-genomes/*.fna | while read line; do name=$(echo $line | cut -d'/' -f2 |cut -d'_' -f4,5,6| cut -d'.' -f1); sh changing-headers.sh $line $name; done
$ ls
capsici_1101.fna insidiosus_R1-1.fna nebraskensis_DOAB_397.fna
capsici_1106.fna insidiosus_R1-3.fna nebraskensis_HF4.fna
capsici_1207.fna nebraskensis_419B.fna nebraskensis_NCPPB_2581.fna
capsici_PF008.fna nebraskensis_44.fna nebraskensis_SL1.fna
Cmm.fna nebraskensis_61-1.fna sepedonicus_ATCC33113.fna
insidiosus_ATCC_10253.fna nebraskensis_7580.fna sepedonicus_CFIA-Cs3N.fna
insidiosus_CFBP_1195.fna nebraskensis_A6096.fna sepedonicus_CFIA-CsR14.fna
insidiosus_CFBP_2404.fna nebraskensis_CFBP_7577.fna tessellarius_ATCC_33566.fna
insidiosus_CFBP_6488.fna nebraskensis_CIBA.fna tessellarius_DOAB_609.fna
insidiosus_LMG_3663.fna nebraskensis_DOAB_395.fna
The outgroups
As I explored the data, I became aware that in the Capsicum and Tuberosum samples, one of the dominant OTUs was Agrobacterium tumefaciens :
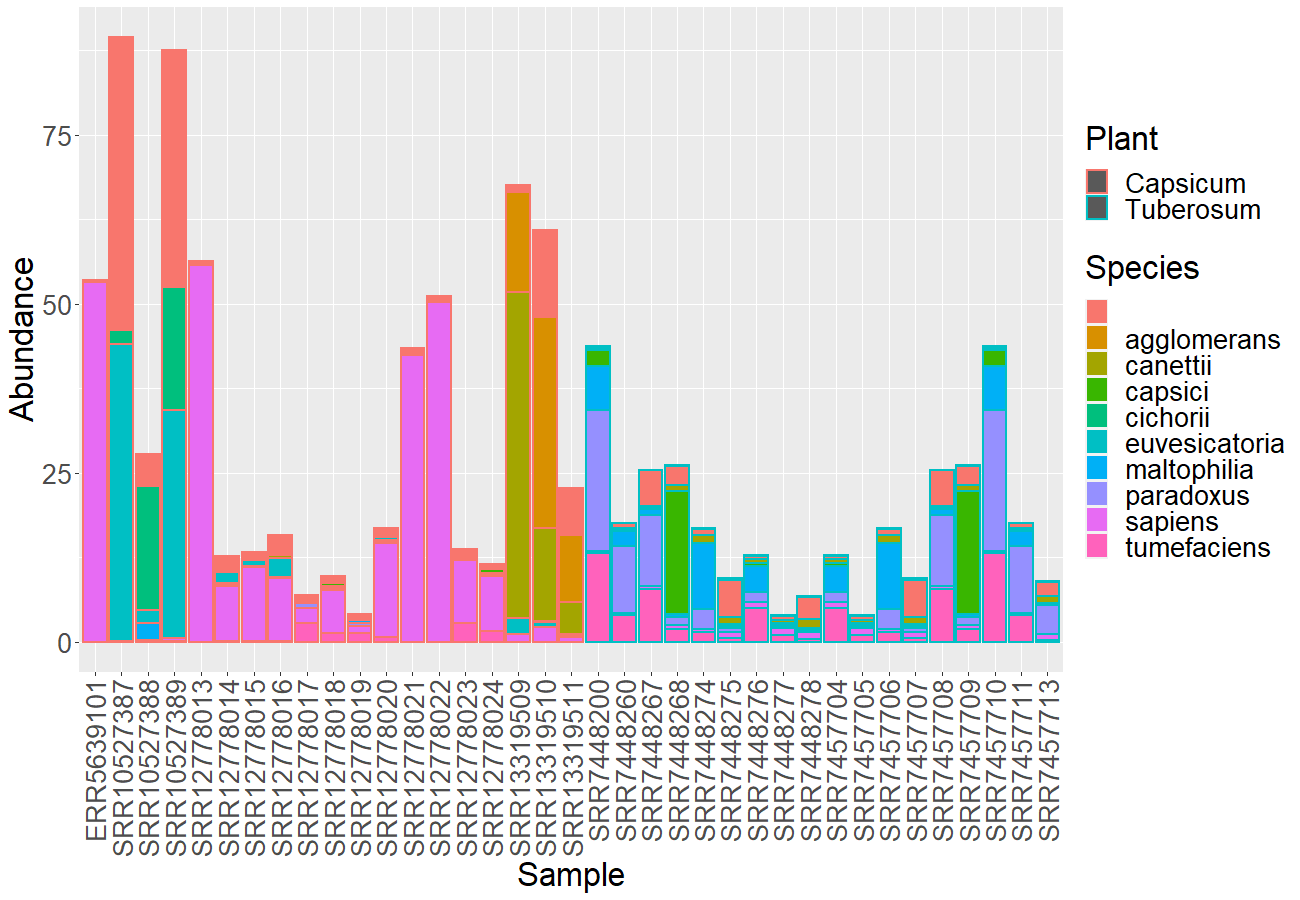
In order to see if this obtained reads do not belong to this dominant population, I add the Agrobacterium tumefaciens to the database.
I will also add an outgroup-like genome from Nostoc desertorum.
With these genomes I will create a Blast database. First I will concatenate all this genomes in one big file and then I will run the next lines of code to do the BLAST database:
$ cat trim-c-genomes/* outgroups/* > all-genomes.fasta
$ cd ../blast
$ makeblastdb -in ../reference/all-genomes.fasta -dbtype nucl -out databases/all-genomes
Dividing the files of individual reads
Now that we have all we need to try a Blast of the “Cmm” extracted-reads, I will
take the Capsicum/choi-2020 data to try this. I will create a directory at the same level
as all-clavi-g/ directory, and I will name it capsi-choi/. Here I will copy
the extracted Cmm from choi-2020/ inside a folder named extracted-cmm-choi/. If we explore one of these files, we will see that is composed for a set of reads:
$ head -n15 extracted-cmm-choi/cmm-capsi-choi-2020-SRR12778013-1.fasta
>SRR12778013.32167716 32167716 length=151
TCGTCATCGACTGCCCCAACCGGCAGGGCGGCCCCCTCACCCTCTCCGCCCTCAACGCCG
CCGACACCGTCGTCTACGCCGCCACCCCCAGCGGCGACGGCGTCGACGGCGTCGCCGGCG
CCCGCCGCACCGTCGACCAGTTCCGGATCAA
>SRR12778013.32168284 32168284 length=151
TCGTCATCGACTGCCCCAACCGGCAGGGCGGCCCCCTCACCCTCTCCGCCCTCAACGCCG
CCGACACCGTCGTCTACGCCGCCACCCCCAGCGGCGACGGCGTCGACGGCGTCGCCGGCG
CCCGCCGCACCGTCGACCAGTTCCGGATCAA
>SRR12778013.32987997 32987997 length=151
TTGTGATCGGAGACCTCGTCGCGCAGCCGGGCGGCCAACTCGAACTTGAGTTCGCCGGCG
GCGATGAGCATCTGGGCGTTGAGATCCTCGATGATCGCCTCGAGCTCGGCACCGCCCGAG
GCGCCCTTCGCGCCCGAGCCCAGCGACGGGG
>SRR12778013.34496501 34496501 length=151
ACGTCGTCGTGTACTGCTGGGGCCCCGGCTGCAACGGCAGCACCCGCGCCGGCCTCGCCC
TCGCGCTCGCGGGCTACGGCCGCGTGAAGGAGCTGGTCGGCGGCTACGAGTACTGGGTCC
In order to separe each sequence in a file, I have prepared a little program
dividing-ind-reads.sh
$ cat dividing-ind-reads.sh
# !/bin/bash
#This program is ment to separate the reads of a fasta file. In order to
#work, the read header must begin with a '>' as the next example:
# >SRR13319511.872821 872821 length=101
#This little program asks for the folder where the fasta files are located
fas=$1 # folder where the fasta files are located
#CREATE A FOLDER TO CONTAIN THE READS
mkdir -p $fas/ind-reads
#EXTRACTING THE INDIVIDUAL READS
ls $fas | grep -v 'ind-reads'| while read line; do name=$(echo $line | cut -d'.' -f1);
file=$(echo $line);
grep '>' $fas/$file | while read line; do ref=$(echo $line| cut -d' ' -f1| cut -d'.' -f2);
grep "$line" $fas/$file -A 3 > ind-temp.fasta;
echo ">"$name-$ref > $name-$ref.fasta;
grep 'G' ind-temp.fasta >> $name-$ref.fasta;
rm ind-temp.fasta;
mv $name-$ref.fasta $fas/ind-reads;
done;done
I will use this program to divide the sequences of each of the fasta files inside
the extracted-cmm-choi/ direcotry:
$ sh dividing-ind-reads.sh extracted-cmm-choi
$ ls extracted-cmm-choi/ind-reads/ | wc -l
7392
Exploration BLAST
Now we are ready to do the BLAST with the mock microbiome that we amassed. I
prepared another little program to do the blast, sec-blast.sh:
$ cat sec-blast.sh
# !/bin/bash
#This program is ment to do a Blast search of a set of files on a database.
# The user needs to specify 1) where is located that database and 2) the
#location of the sequences that are going to be submitted to blast
db=$1 #Location of the database to do the blast
seq=$2 #Location of the sequences to blast
mkdir output-blast
ls $seq | while read line; do name=$(echo $line | cut -d'.' -f1);
for i in 0.000001 ; do mkdir -p output-blast/$i;
blastn -db $db -query $seq/$line -outfmt "6 sseqid slen qstart qend bitscore evalue sseq " -evalue $i -num_threads 12 -out output-blast/$i/$name;
done;done
It is important to note that one of the advantages of the -outfmt 6 of BLAST is
that one can customize the output . We are asking for 6 parameters:
- sseqid: Subject Seq-id
- slen: Subject sequence length
- qstart: Start of alignment in query
- qend: End of alignment in query
- bitscore: Bit score
- evalue: Expect value
- sseq: Aligned part of subject sequence
$ sh sec-blast.sh ../databases/all-genomes extracted-cmm-choi/ind-reads/
It will take a long time since we got a great amount of sequences to process. At the end, if we take a look at how many of the 7392 sequences got a result, we will see that more than the 75% got no output after the BLAST:
$ ls -l output-blast/0.000001/ | grep ' 0 ' | wc -l
5850
If I take some of these sequences with no output and sumbit them to a BLAST process with all the NCBI database, I find insteresting results. Some show a high identity and good e-value with other Cmm lineajes that I do not have in the dabase (we only put one Cmm of reference). While others, show this tendency against plasmid sequences:
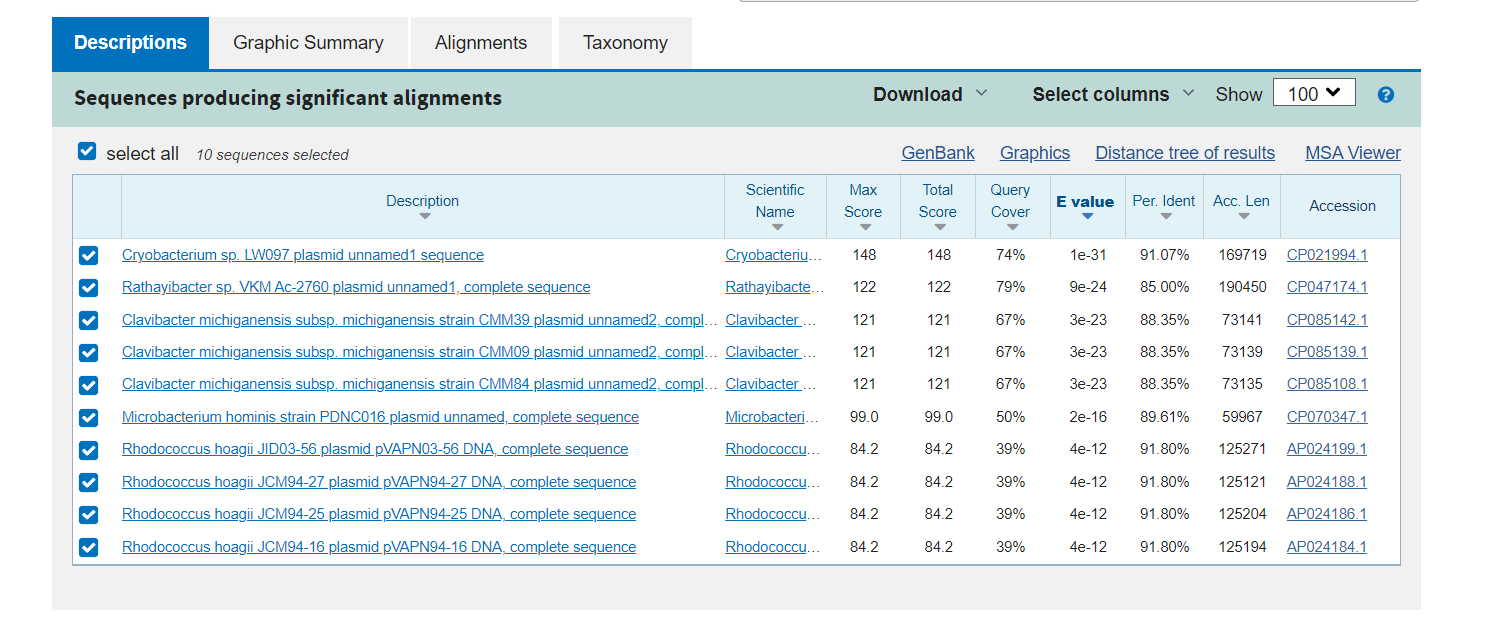
Adjusting the BLAST database
With this information, I searched for a new set of sequences:
| Entity | Number of sequences |
|---|---|
| Cmm | 10 |
| Plasmids | 5 |
I obtained the Cmm genomes from the NCBI repository
In order to trim their headers, I enhanced (at least I think is a better program),
the changing-headers.sh script and did a new program in head-genomes-trim.sh.
This program allows to change a great amount of genomes with only one line of code:
$ cat head-genomes-trim.sh
#!/bin/sh
# This program is to change the headers of a fasta file in order for all the
#headers to be the same for the entire file
# This is useful when the user want to concatenate all the genomes from a
#database and want to know from which of the initial genomes is the
#best match
#This program ask from the user to specify 1 input: 1) the location
#of the fasta files without the "/" character at the end
gem=$1 #Location to the genomes in fasta format without the "/" character at the end
# CREATE A DIRECTORY TO STORE THE TRIMMED GENOME
mkdir -p $gem/trim-header-genomes
ls $gem | grep -v 'trim-header-genomes' | while read line;
do fas=$(echo $line);
#CREATE A .TXT FILE TO STORE THE ORIGINAL HEADERS
grep '>' $gem/$fas > heads.txt;
cp $gem/$fas $gem/trim-header-genomes/htrim-$fas;
#REPLACE ALL THE HEADERS FOR THE SUFFIX NAME
cat heads.txt | while read line;
do sed "s/$line/>$fas/" $gem/trim-header-genomes/htrim-$fas > $gem/trim-header-genomes/temp.txt;
mv $gem/trim-header-genomes/temp.txt $gem/trim-header-genomes/htrim-$fas;
done;done
#REMOVING TEMP FILES
rm heads.txt
After submitting the new sequences to this program. I will do a new BLAST database to see if the number of identified sequences is better:
$ cat trim-c-genomes/* outgroups/* plasmids/trim-header-genomes/* cmm/other-cmm/trim-header-genomes/* > enhanced-datab.fasta
$ makeblastdb -in enhanced-datab.fasta -dbtype nucl -out ../blast/databases/enhnced-datab
With this assembled, we will run again the BLAST with this new database. First, I
will move the first output to a new folder so as to not get overwritten. I will use
again the sec-blast.sh.
$ mv output-blast/ first-database-output/
$ sh sec-blast.sh ../databases/enhnced-datab extracted-cmm-choi/ind-reads/
Again, since we have a lot of sequences, it will take some minutes to complete the process. If we want to see the results, we will see that close to 400 sequences are now identified. But we still have more than 50% of the sequences without an output:
$ ls -l capsi-choi/output-blast/0.000001/ | grep ' 0 ' | wc -l
5431
BLAST with the plasmid sequences
It is interesting that some of the sequences presented a better e-value when aligned to a plasmid sequence. I will do a database with these sequences and see how many of the sequences get an output.
$ cat plasmids/trim-header-genomes/* > plasmids.fasta
$ makeblastdb -in ../reference/plasmids.fasta -dbtype nucl -out ../blast/databases/plasmids
$ sh sec-blast.sh ../databases/plasmids extracted-cmm-choi/ind-reads/
$ ls -l output-blast/0.000001/ | grep -v ' 0 ' | wc -l
11
There are not as much as I expected, but the results are quite interesting and worth noting them. In this example, I only added 5 sequences of plasmids and 10 Cmm lineajes. Indeed, the results were better that the former database, but more than 70% of the sequences (only from Choi 2020) are still unidentified. Certainly, using nucleotides usially brings less outputs than a “faraway” proximation as using aminoacids. But this result suggests that across Cmm lineajes there are a great amount of nucleic substitutions. Evaluate if they are synonymous or nonsynonymous can be a good approach to follow.