clavibacter
Extraction of Clavibacter reads

![]()
The focus of this project is to obtain and analize the diversity of Clavibacter lineages living with the already mentioned plants.
I am going to use the kraken-tools
set of scripts. Specifically, I will use the extract_kraken_reads.py script to obtain
specific reads from the entire fastq files.
Exploring the extract_kraken_reads.py script
First, I will explore the options that the use extract_kraken_reads.py script.
$ extract_kraken_reads.py --help
usage: extract_kraken_reads.py [-h] -k KRAKEN_FILE -s SEQ_FILE1
[-s2 SEQ_FILE2] -t TAXID [TAXID ...] -o
OUTPUT_FILE [-o2 OUTPUT_FILE2] [--append]
[--noappend] [--max MAX_READS] [-r REPORT_FILE]
[--include-parents] [--include-children]
[--exclude] [--fastq-output]
optional arguments:
-h, --help show this help message and exit
-k KRAKEN_FILE Kraken output file to parse
-s SEQ_FILE1, -s1 SEQ_FILE1, -1 SEQ_FILE1, -U SEQ_FILE1
FASTA/FASTQ File containing the raw sequence letters.
-s2 SEQ_FILE2, -2 SEQ_FILE2
2nd FASTA/FASTQ File containing the raw sequence
letters (paired).
-t TAXID [TAXID ...], --taxid TAXID [TAXID ...]
Taxonomy ID[s] of reads to extract (space-delimited)
-o OUTPUT_FILE, --output OUTPUT_FILE
Output FASTA/Q file containing the reads and sample
IDs
-o2 OUTPUT_FILE2, --output2 OUTPUT_FILE2
Output FASTA/Q file containig the second pair of reads
[required for paired input]
--append Append the sequences to the end of the output FASTA
file specified.
--noappend Create a new FASTA file containing sample sequences
and IDs (rewrite if existing) [default].
--max MAX_READS Maximum number of reads to save [default: 100,000,000]
-r REPORT_FILE, --report REPORT_FILE
Kraken report file. [required only if --include-
parents/children is specified]
--include-parents Include reads classified at parent levels of the
specified taxids
--include-children Include reads classified more specifically than the
specified taxids
--exclude Instead of finding reads matching specified taxids,
finds all reads NOT matching specified taxids
--fastq-output Print output FASTQ reads [requires input FASTQ,
default: output is FASTA]
So we will need 4 main inputs:
- -k [I will indicate where the kraken file is located]
- -s and -s2 [The location where the reads forward and reverse are located]
- -t [The taxonomic ID that is assignated to each lineaje by kraken ]
- -o and -o2 [The location and name of the output files]
The tax-id can be located on the kraken.report file. In the next example, the
tax-id is highlited by a blood-red square:
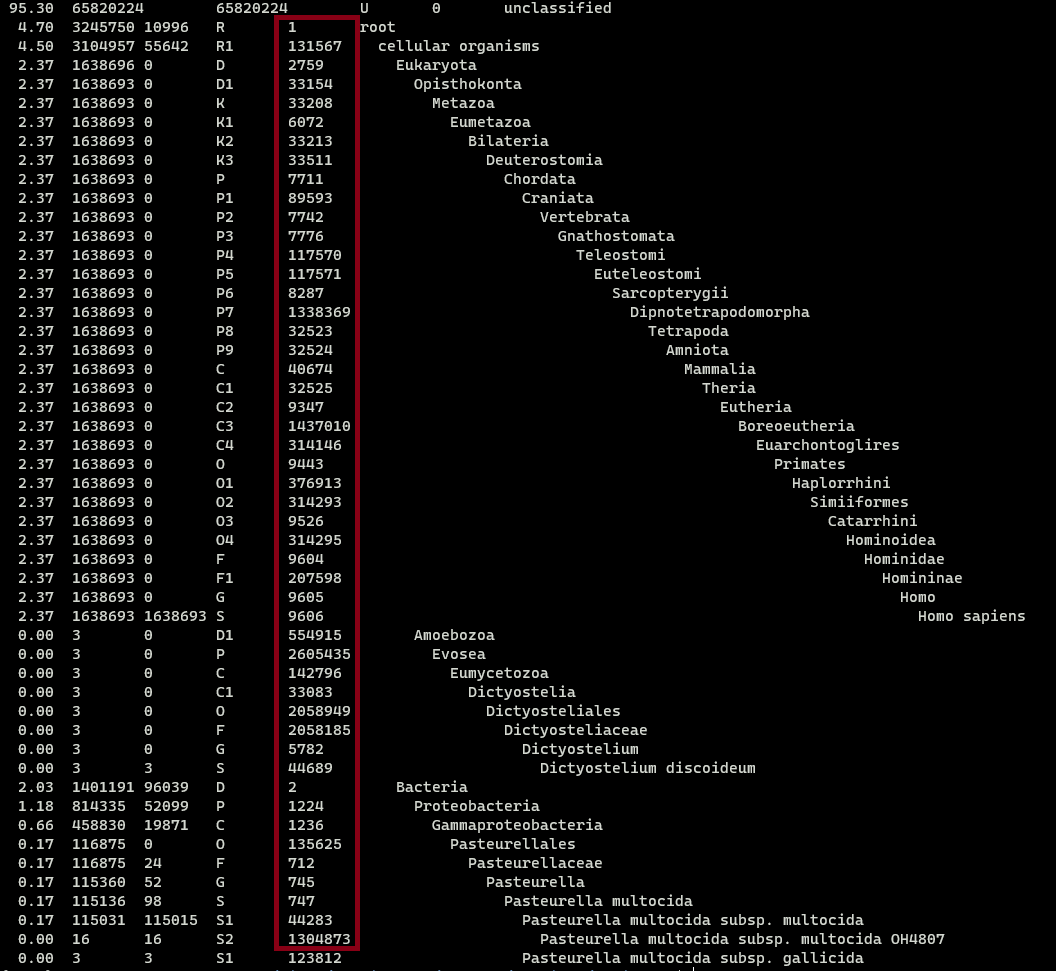
Extracting Clavibacter reads
I will use as an example the data located in capsicum/choi-2020/ folder. Let’s
remember what is inside the reads/ folder:
$ ls -1 reads/
SRR12778013-1.fastq
SRR12778013-2.fastq
SRR12778014-1.fastq
SRR12778014-2.fastq
SRR12778015-1.fastq
SRR12778015-2.fastq
SRR12778016-1.fastq
SRR12778016-2.fastq
SRR12778017-1.fastq
SRR12778017-2.fastq
SRR12778018-1.fastq
SRR12778018-2.fastq
SRR12778019-1.fastq
SRR12778019-2.fastq
SRR12778020-1.fastq
SRR12778020-2.fastq
SRR12778021-1.fastq
SRR12778021-2.fastq
SRR12778022-1.fastq
SRR12778022-2.fastq
SRR12778023-1.fastq
SRR12778023-2.fastq
SRR12778024-1.fastq
SRR12778024-2.fastq
I will use the first set of reads SRR12778013 to run an example. I will create a
clavi/ folder inside the reads/ folder to allocate the fastq outputs of the
extraction. Now, with the next line, I will extract Clavibacter reads from this
library:
$ extract_kraken_reads.py -k taxonomy/kraken/krakens/SRR12778013.kraken -r taxonomy/kraken/reports/SRR12778013.report -s1 reads/SRR12778013-1.fastq -s2 reads/SRR12778013-2.fastq -o reads/clavi/capsi-choi-2020-SRR12778013-clav-1.fq -o2 reads/clavi/capsi-choi-2020-SRR12778013-clav-2.fq -t 1573 --fastq-output --include-children
PROGRAM START TIME: 05-06-2022 17:33:10
1 taxonomy IDs to parse
>> STEP 1: PARSING KRAKEN FILE FOR READIDS taxonomy/kraken/krakens/SRR12778013.kraken
69.07 million reads processed
84 read IDs saved
>> STEP 2: READING SEQUENCE FILES AND WRITING READS
84 read IDs found (68.66 mill reads processed)
84 read IDs found (68.66 mill reads processed)
84 reads printed to file
Generated file: reads/clavi/SRR12778013-clav-1.fq
Generated file: reads/clavi/SRR12778013-clav-2.fq
PROGRAM END TIME: 05-06-2022 18:44:52
By the end of the process, we will have a new pair of files that contain the
reads of all the Clavibacter lineajes found, as indicated by the output of
kraken2.
I have prepared a little script that will help to extract all the Clavibacter reads from a set of libraries. It is located on the scripts folder and is called clavi-extract.sh. I will show what is inside it:
$ cat clavi-extract.sh
#!/bin/sh
# This program is going to extract the Clavibacter reads from a set of samples.
# You need to specify the program 1)the name of the author or a first prefix to name
# the output files, and 2) a second prefix that is advised to be the name of the host plant.
aut=$1 #A prefix to name some of the files. In this case, the author name.
pref=$2 #The first prefix. In this case the name of the plant's host
# EXTRACTING THE CLAVIBACTER READS
# With the next piece of code, the reads clasiffied as from the genus "Clavibacter", will be separated from the main reads.
# The number needed for the extraction is the numeric-ID given to Clavibacter by kraken2: 1573
# MAKING THE NECESSARY FOLDERS TO ALLOCATE THE OUTPUTS
mkdir -p reads/clavi/
mkdir -p reads/fasta-clavi
#EXTRACT THE READS IN FASTQ FORMAT
cat metadata/run-labels.txt | while read line;
do echo "\nExtracting Clavibacter reads in fastq from sample:" $line;
extract_kraken_reads.py -k taxonomy/kraken/krakens/$line.kraken -r taxonomy/kraken/reports/$line.report -s1 reads/$line-1.fastq -s2 reads/$line-2.fastq -o reads/clavi/$pref-$aut-$line-clav-1.fq -o2 reads/clavi/$pref-$aut-$line-clav-2.fq -t 1573 --fastq-output --include-children; done
#EXTRACT THE READS IN FASTA FORMAT
cat metadata/run-labels.txt | while read line;
do echo "\nExtracting Clavibacter reads in fasta from sample:" $line;
extract_kraken_reads.py -k taxonomy/kraken/krakens/$line.kraken -r taxonomy/kraken/reports/$line.report -s1 reads/$line-1.fastq -s2 reads/$line-2.fastq -o reads/fasta-clavi/$pref-$aut-$line-clav-1.fasta -o2 reads/fasta-clavi/$pref-$aut-$line-clav-2.fasta -t 1573 --include-children; done
This will take all the reads that were downloaded and which identification ids are
on the run-labels.txt file. It is important to note that the script needs two
prefixes as input. It can take several minutes to finish to process all
the reads. The second part of the script is going to extract the reads but the
output will be in fasta format, and will be locaten inside the fasta-clavi/
folder. In the end, we will have the complete extracted reads on fastq format on the clavi/ folder and on fasta format on fasta-clavi/:
$ sh clavi-extract.sh choi-2020 capsi
$ for i in clavi fasta-clavi; do echo -e '\n'$i ; ls reads/$i; done
clavi
capsi-choi-2020-SRR12778013-clav-1.fq capsi-choi-2020-SRR12778019-clav-2.fq
capsi-choi-2020-SRR12778013-clav-2.fq capsi-choi-2020-SRR12778020-clav-1.fq
capsi-choi-2020-SRR12778014-clav-1.fq capsi-choi-2020-SRR12778020-clav-2.fq
capsi-choi-2020-SRR12778014-clav-2.fq capsi-choi-2020-SRR12778021-clav-1.fq
capsi-choi-2020-SRR12778015-clav-1.fq capsi-choi-2020-SRR12778021-clav-2.fq
capsi-choi-2020-SRR12778015-clav-2.fq capsi-choi-2020-SRR12778022-clav-1.fq
capsi-choi-2020-SRR12778016-clav-1.fq capsi-choi-2020-SRR12778022-clav-2.fq
capsi-choi-2020-SRR12778016-clav-2.fq capsi-choi-2020-SRR12778023-clav-1.fq
capsi-choi-2020-SRR12778017-clav-1.fq capsi-choi-2020-SRR12778023-clav-2.fq
capsi-choi-2020-SRR12778017-clav-2.fq capsi-choi-2020-SRR12778024-clav-1.fq
capsi-choi-2020-SRR12778018-clav-1.fq capsi-choi-2020-SRR12778024-clav-2.fq
capsi-choi-2020-SRR12778018-clav-2.fq SRR12778013-clav-1.fq
capsi-choi-2020-SRR12778019-clav-1.fq SRR12778013-clav-2.fq
fasta-clavi
capsi-choi-2020-SRR12778013-clav-1.fasta capsi-choi-2020-SRR12778019-clav-1.fasta
capsi-choi-2020-SRR12778013-clav-2.fasta capsi-choi-2020-SRR12778019-clav-2.fasta
capsi-choi-2020-SRR12778014-clav-1.fasta capsi-choi-2020-SRR12778020-clav-1.fasta
capsi-choi-2020-SRR12778014-clav-2.fasta capsi-choi-2020-SRR12778020-clav-2.fasta
capsi-choi-2020-SRR12778015-clav-1.fasta capsi-choi-2020-SRR12778021-clav-1.fasta
capsi-choi-2020-SRR12778015-clav-2.fasta capsi-choi-2020-SRR12778021-clav-2.fasta
capsi-choi-2020-SRR12778016-clav-1.fasta capsi-choi-2020-SRR12778022-clav-1.fasta
capsi-choi-2020-SRR12778016-clav-2.fasta capsi-choi-2020-SRR12778022-clav-2.fasta
capsi-choi-2020-SRR12778017-clav-1.fasta capsi-choi-2020-SRR12778023-clav-1.fasta
capsi-choi-2020-SRR12778017-clav-2.fasta capsi-choi-2020-SRR12778023-clav-2.fasta
capsi-choi-2020-SRR12778018-clav-1.fasta capsi-choi-2020-SRR12778024-clav-1.fasta
capsi-choi-2020-SRR12778018-clav-2.fasta capsi-choi-2020-SRR12778024-clav-2.fasta
Now, we can use this reads for future analysis.
Extracting Clavibacter michiganensis michiganensis reads
If I explore the diversity of Clavibacter lineajes inside each one of the
libraries, it can be seen that there is a variety of lineajes that have been
identified. For example, this is the output from the two first libraries from the
choi-2020 dataset:
$ for i in 3 4; do echo -e "\n"Clavibacter lineajes identified in library SRR1277801$i; grep 'Clavibacter' taxonomy/kraken/reports/SRR1277801$i.report; done
Clavibacter lineajes identified in library SRR12778013
0.00 544 84 G 1573 Clavibacter
0.00 398 151 S 28447 Clavibacter michiganensis
0.00 62 62 S1 1874630 Clavibacter michiganensis subsp. capsici
0.00 49 49 S1 31965 Clavibacter michiganensis subsp. tessellarius
0.00 33 33 S1 31964 Clavibacter michiganensis subsp. sepedonicus
0.00 32 32 S1 31963 Clavibacter michiganensis subsp. nebraskensis
0.00 29 29 S1 1401995 Clavibacter michiganensis subsp. californiensis
0.00 25 25 S1 33014 Clavibacter michiganensis subsp. insidiosus
0.00 17 15 S1 33013 Clavibacter michiganensis subsp. michiganensis
0.00 2 2 S2 443906 Clavibacter michiganensis subsp. michiganensis NCPPB 382
0.00 34 34 S 2768071 Clavibacter zhangzhiyongii
0.00 28 0 G1 2626594 unclassified Clavibacter
0.00 28 28 S 2860285 Clavibacter sp. A6099
Clavibacter lineajes identified in library SRR12778014
0.03 20092 3466 G 1573 Clavibacter
0.02 13883 5266 S 28447 Clavibacter michiganensis
0.00 2159 2159 S1 1874630 Clavibacter michiganensis subsp. capsici
0.00 1912 1912 S1 31965 Clavibacter michiganensis subsp. tessellarius
0.00 1161 1161 S1 31964 Clavibacter michiganensis subsp. sepedonicus
0.00 977 977 S1 31963 Clavibacter michiganensis subsp. nebraskensis
0.00 921 921 S1 1401995 Clavibacter michiganensis subsp. californiensis
0.00 787 787 S1 33014 Clavibacter michiganensis subsp. insidiosus
0.00 679 647 S1 33013 Clavibacter michiganensis subsp. michiganensis
0.00 32 32 S2 443906 Clavibacter michiganensis subsp. michiganensis NCPPB 382
0.00 21 21 S1 1734031 Clavibacter michiganensis subsp. phaseoli
0.00 1935 1935 S 2768071 Clavibacter zhangzhiyongii
0.00 808 0 G1 2626594 unclassified Clavibacter
0.00 808 808 S 2860285 Clavibacter sp. A6099
0.00 1 0 S 2560388 Clavibacter virus CN1A
0.00 1 1 S1 1406793 Clavibacter phage CN1A
0.00 1 0 S 2560387 Clavibacter virus CMP1
0.00 1 1 S1 686439 Clavibacter phage CMP1
There are reads from Clavibacter michiganensis michiganensis (Cmm) identified. I have
prepared a script to extract this reads from all the libraries in a folder. It is
called ccm-extract.sh and can be located on the on the scripts folder.
$ cat cmm-extract.sh
#!/bin/sh
# This program is going to extract the Clavibacter michiganensis michiganensis reads from a set of samples
# The program ask you to give two pieces of information:
# 1) A first prefix that is the name of the author were the data has been extracted
# 2) A second prefix that is the name of the host plant (in the case of the project were this script was created)
aut=$1 #A prefix to name some of the files. In this case, the author name.
pref=$2 #The first prefix. In this case the name of the plant's host
# CREATING NEEDED FOLDERS
mkdir -p reads/cmm/
# EXTRACTING THE CLAVIBACTER MICHIGANESIS-MICHIGANENSIS READS
# With the next piece of code, the reads clasiffied as from the Clavibacter michiganensis michiganensis, will be separated from the main reads.
# The number needed for the extraction is the numeric-ID given to Cmm by kraken2: 33013
cat metadata/run-labels.txt | while read line;
do echo "\nExtracting Cmm reads in fasta format from sample:" $line; extract_kraken_reads.py -k taxonomy/kraken/krakens/$line.kraken -r taxonomy/kraken/reports/$line.report -s1 reads/$line-1.fastq -s2 reads/$line-2.fastq -o reads/cmm/cmm-$pref-$aut-$line-1.fasta -o2 reads/cmm/cmm-$pref-$aut-$line-2.fasta -t 33013 --include-children; done
Again, we need the Cmm tax-id that is 33013 and to specify the two prefixes as input to run the script.
$ sh cmm-extract.sh choi-2020 capsi
$ ls reads/cmm/
cmm-capsi-choi-2020-SRR12778013-1.fasta cmm-capsi-choi-2020-SRR12778019-1.fasta
cmm-capsi-choi-2020-SRR12778013-2.fasta cmm-capsi-choi-2020-SRR12778019-2.fasta
cmm-capsi-choi-2020-SRR12778014-1.fasta cmm-capsi-choi-2020-SRR12778020-1.fasta
cmm-capsi-choi-2020-SRR12778014-2.fasta cmm-capsi-choi-2020-SRR12778020-2.fasta
cmm-capsi-choi-2020-SRR12778015-1.fasta cmm-capsi-choi-2020-SRR12778021-1.fasta
cmm-capsi-choi-2020-SRR12778015-2.fasta cmm-capsi-choi-2020-SRR12778021-2.fasta
cmm-capsi-choi-2020-SRR12778016-1.fasta cmm-capsi-choi-2020-SRR12778022-1.fasta
cmm-capsi-choi-2020-SRR12778016-2.fasta cmm-capsi-choi-2020-SRR12778022-2.fasta
cmm-capsi-choi-2020-SRR12778017-1.fasta cmm-capsi-choi-2020-SRR12778023-1.fasta
cmm-capsi-choi-2020-SRR12778017-2.fasta cmm-capsi-choi-2020-SRR12778023-2.fasta
cmm-capsi-choi-2020-SRR12778018-1.fasta cmm-capsi-choi-2020-SRR12778024-1.fasta
cmm-capsi-choi-2020-SRR12778018-2.fasta cmm-capsi-choi-2020-SRR12778024-2.fasta
Adjusting the all-aroun program
Finally, I will add this step to the programm that I have been constructing along these episodes.
$ cat kraken-ext.sh
#!/bin/sh
# This is a program that is going to pick a SraRunTable of metadata and
#extract the run label to download, trim and move the libraries information.
# This program requires that you give 2 input data: 1) where this
#SraRunTable is located, 2) where the kraken database has been saved,
# 3) the name of the author of the library, and 4) A prefix for some of the
# output files, in this case the name of the plant's host.
#ASSIGNATIONS
metd=$1 #Location to the SraRunTable.txt
kdat=$2 #Location of the kraken2 database
aut=$3 #A first prefix to name some of the files. In this case, the author name.
pref=$4 #The second prefix. In this case the name of the plant's host
root=$(pwd) #Gets the path to the directory of this file, on which the outputs ought to be created
# Now we will define were the reads are:
runs='reads'
# CREATING NECCESARY FOLDERS
mkdir reads
mkdir -p taxonomy/kraken
mkdir -p taxonomy/taxonomy-logs/scripts
mkdir -p taxonomy/kraken/reports
mkdir -p taxonomy/kraken/krakens
mkdir -p taxonomy/biom-files
mkdir -p reads/clavi/
mkdir -p reads/cmm/
mkdir -p reads/fasta-clavi
# DOWNLOADING THE DATA
#Let's use the next piece of code to download the data
cat $metd | sed -n '1!p' | while read line; do read=$(echo $line | cut -d',' -f1); fasterq-dump -S $read -p -e 8 -o $read ; done
mv *.fastq reads/
# The -e flag can be customized. This indicates the number of threads used to do this task.
# MANAGING THE DOWNLADED DATA
# We will change the names of the reads files. They have a sufix that makes impossible
#to be read in a loop
ls $runs | while read line ; do new=$(echo $line | sed 's/_/-/g'); mv $runs/$line $runs/$new; done
# Now, we will create a file where the information of the run labes can be located
cat $metd | sed -n '1!p' | while read line; do read=$(echo $line | cut -d',' -f1); echo $read ; done > run-labels.txt
mv run-labels.txt metadata/
# TAXONOMIC ASSIGNATION WITH KRAKEN2
cat metadata/run-labels.txt | while read line; do file1=$(echo $runs/$line-1.fastq); file2=$(echo $runs/$line-2.fastq) ; echo '\n''working in run' "$line"\
#kraken2 --db $kdat --threads 6 --paired $file1 $file2 --output taxonomy/kraken/krakens/$line.kraken --report taxonomy/kraken/reports/$line.report \
echo '#!/bin/sh''\n''\n'"kraken2 --db $kdat --threads 6 --paired" "$runs/$line"'-1.fastq' "$runs/$line"'-2.fastq' "--output taxonomy/kraken/krakens/$line.kraken --report taxonomy/kraken/reports/$line.report" > taxonomy/taxonomy-logs/scripts/$line-kraken.sh; sh taxonomy/taxonomy-logs/scripts/$line-kraken.sh; done
#CREATING THE BIOM FILE
# Now we will create the biom file using kraken-biom
kraken-biom taxonomy/kraken/reports/* --fmt json -o taxonomy/biom-files/$aut.biom
# EXTRACTING THE CLAVIBACTER READS
# With the next piece of code, the reads clasiffied as from the genus "Clavibacter", will be separated from the main reads.
# The number needed for the extraction is the numeric-ID given to Clavibacter by kraken2: 1573
#EXTRACT THE READS IN FASTQ FORMAT
cat metadata/run-labels.txt | while read line;
do echo "\nExtracting Clavibacter reads in fastq from sample:" $line;
extract_kraken_reads.py -k taxonomy/kraken/krakens/$line.kraken -r taxonomy/kraken/reports/$line.report -s1 reads/$line-1.fastq -s2 reads/$line-2.fastq -o reads/clavi/$pref-$aut-$line-clav-1.fq -o2 reads/clavi/$pref-$aut-$line-clav-2.fq -t 1573 --fastq-output --include-children; done
#EXTRACT THE READS IN FASTA FORMAT
cat metadata/run-labels.txt | while read line;
do echo "\nExtracting Clavibacter reads in fasta from sample:" $line;
extract_kraken_reads.py -k taxonomy/kraken/krakens/$line.kraken -r taxonomy/kraken/reports/$line.report -s1 reads/$line-1.fastq -s2 reads/$line-2.fastq -o reads/fasta-clavi/$pref-$aut-$line-clav-1.fasta -o2 reads/fasta-clavi/$pref-$aut-$line-clav-2.fasta -t 1573 --include-children; done
# EXTRACTING THE CLAVIBACTER MICHIGANESIS-MICHIGANENSIS READS
# With the next piece of code, the reads clasiffied as from the Clavibacter michiganensis michiganensis, will be separated from the main reads.
# The number needed for the extraction is the numeric-ID given to Cmm by kraken2: 33013
cat metadata/run-labels.txt | while read line;
do echo "\nExtracting Cmm reads in fasta format from sample:" $line; extract_kraken_reads.py -k taxonomy/kraken/krakens/$line.kraken -r taxonomy/kraken/reports/$line.report -s1 reads/$line-1.fastq -s2 reads/$line-2.fastq -o reads/cmm/cmm-$pref-$aut-$line-1.fasta -o2 reads/cmm/cmm-$pref-$aut-$line-2.fasta -t 33013 --include-children; done
With this modification, we can automatically do all the bash steps that we have
covered in the last episodes.